









Article / Short Article
1</supSotiria hospital for thoracic diseases,
2Kifisia institution of profesional training
Efstathios Koutsostathis,
Kerameikos Health Center,
Greece.
14 November 2023; 2 December 2023
Amyotrophic (poor muscle nourishment)
Lateral (areas of the human spinal cord where nerve cells responsible for muscle control are located)
Sclerosis (destruction and hardening of these areas)
 Dendrites, Soma (cell body), Axon, Nucleus of Schwann cell, motor neuron, muscle
Dendrites, Soma (cell body), Axon, Nucleus of Schwann cell, motor neuron, muscle
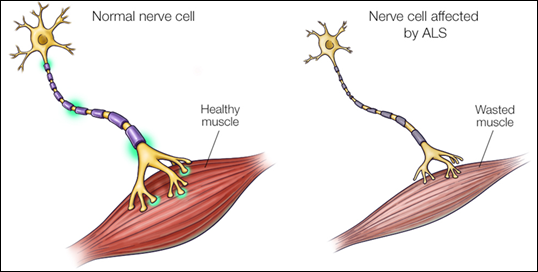
Amyotrophic lateral sclerosis (abbreviated ALS, also referred to as Lou Gehrig’s disease) is a type of motor neuron disease. ALS is the 3rd most common neurodegenerative disorder, after Alzheimer’s and Parkinson’s. The incidence is 1-2 per 100,000 people per year. The main age of onset is 40 to 60 years old. ALS is a progressive, fatal, neurodegenerative disease caused by the degeneration of motor neurons, the nerve cells in the central nervous system that control voluntary muscle movement. The disorder causes muscle weakness and atrophy throughout the body as both the upper and lower motor neurons degenerate, ceasing to send messages to the muscles. Unable to function, the muscles gradually weaken, developing fasciculations due to denervation and eventually atrophy. The patient may eventually lose the ability to initiate and control all voluntary movements. The bladder and bowel sphincters and the muscles responsible for eye movement are usually (but not always) spared.
Early symptoms of ALS include:
- Difficulty lifting the front of the foot and toes.
- Weakness in the lower limbs and the extremities of the feet.
- Hand weakness or clumsiness.
- Unclear articulacy (stuttering) or problems swallowing.
Muscle cramps and nerve contractions in the hands, shoulders and tongue.
Regardless of the part of the body first affected by the disease, muscle weakness and atrophy spread to other parts of the body as the disease progresses. Patients experience increasing difficulty in moving, swallowing (dysphagia) and speaking or forming words (dysarthria).
As the diaphragm and intercostal muscles weaken, vital capacity and inspiratory pressure decrease. Bilevel positive pressure ventilation (often referred to by the trade name BiPAP) is often used to support breathing, first at night and later during the day. It is recommended well before BiPAP becomes inadequate for patients to decide whether to have a tracheostomy and undergo mechanical ventilation. At this point, some patients opt for palliative care.
In the latter stage of the disease, inability to swallow leads to aspiration and increases the risk of aspiration pneumonia. The person becomes malnourished due to dysphagia and weakness of the oesophageal muscles. In this case a gastrostomy is needed. Finally, the respiratory muscles are affected and the patient is led to death. Most people with ALS die from respiratory failure or pneumonia. Death usually occurs within two to five years of diagnosis.
Since familial ALS has been linked to a mutation in the gene encoding superoxide dismutase, a critical enzyme involved in protecting mitochondria from oxidative stress, the initial theory focused on oxidative stress, a possible but non-specific mechanism of almost all neurodegenerative disorders. The mutated SOD1 gene is responsible for causing ALS in a subset of the 10% of all ALS patients who have a familial or genetic form of the disease.
There is one known hereditary factor in familial ALS (FALS).
However, there is no known hereditary component in the 90-95% of cases diagnosed as sporadic ALS.
The children of those diagnosed with familial ALS have a higher risk factor for developing the disease of 50%. However, those with close family members diagnosed with sporadic ALS do not have a higher risk factor than the general population, suggesting an environmental or other non-genetic cause.
A number of environmental etiological factors have been suggested for the increased incidence in the Western Pacific. Prolonged exposure to a dietary neurotoxin called BMAA is a suspected risk factor in Guam. The neurotoxin is a compound found in the seed of Cycas circinalis, a tropical plant found in Guam that was used in human nutrition in the 1950s and early 1960s.
The very high incidence of the disease among Italian footballers (over five times higher than usually expected) has raised concern about a possible link between the disease and the use of pesticides on football pitches (several of which have been linked to neuronal toxicity).
According to the ALS Association, military veterans are at increased risk of ALS (again, possibly implying a link to exposure to neurotoxic chemicals). In its report ALS in the Military, the group noted a nearly 60% greater chance of developing the disease in military veterans than in the general population.
For Gulf War veterans, the likelihood is considered twice as high as that of non-deployed Gulf War veterans in a joint study by the Veterans Affairs Administration and the DOD, another epidemiological association that suggests a link to toxic exposure.
Dietary intake of polyunsaturated fatty acids (PUFAs) has been shown in several studies to reduce the risk of developing ALS and other neurodegenerative disorders.
No single test can give a definitive diagnosis of ALS, although the presence of upper and lower motor neuron signs in a single limb is strongly suggestive. Instead, the diagnosis of ALS is based primarily on the symptoms and signs observed by the doctor in the patient and on a series of tests to rule out other diseases.
Doctors take the patient’s complete medical history and usually conduct neurological examinations at regular intervals to assess whether symptoms such as muscle weakness, muscle atrophy, hyperreflexia and spasticity are progressively worsening.
Because the symptoms of ALS may be similar to those of a wide variety of other diseases or disorders, appropriate tests should be performed to rule out this possibility.
One of these tests is electromyography (EMG), a special recording technique that detects electrical activity in the muscles. Some EMG findings can support the diagnosis of ALS.
Another common test measures nerve conduction velocity (NCV). Specific abnormalities in NCV results may suggest, for example, that the patient has a form of peripheral neuropathy (damage to peripheral nerves) or myopathy (muscle disease) instead of ALS.
The doctor on the MRI scan can see detailed images of the brain and spinal cord. Based on the patient’s symptoms and findings from the exam and from these tests, the doctor may order tests on blood and urine samples to eliminate the possibility of other diseases, as well as laboratory tests. In some cases, for example, if a doctor suspects that a patient may have myopathy rather than ALS, a muscle biopsy may be performed.
The Food and Drug Administration (FDA) approved the first drug treatment for the disease: Riluzole (Rilutek). Clinical trials with ALS patients have shown that Riluzole prolongs survival by several months and may have a greater survival benefit for those with bulbar onset. The drug also extends the time before the patient needs ventilatory support. Patients taking the drug should be monitored for liver damage and other possible side effects.
Other treatments for ALS are designed to relieve symptoms and improve patients’ quality of life. This supportive care is best provided by multidisciplinary teams of health professionals: such as doctors, physical therapists, occupational therapists, speech and language therapists, nutritionists, social workers, and home health nurses. Respiratory support and assistive devices for speech and movement.
Life for people with ALS is particularly difficult and, especially in the final stages of the disease, ongoing care and support from family and health professionals is essential.
Working with patients and caregivers, these teams can design an individualized medical and physical therapy plan and provide special equipment to keep patients as mobile and comfortable as possible.
Doctors can prescribe medications to help reduce fatigue, reduce muscle cramps, control spasticity and reduce excess saliva and phlegm. Medications are also available to help patients with pain, depression, sleep disorders and constipation. This is particularly useful in terms of patients with dysphagia, which many ALS patients have experienced.
- https://www.diaplasis.eu/%CE%B1%CE%BC%CF%85%CE%BF%CF%84%CF%81%CE%BF%CF%86%CE%B9%CE%BA%CE%AE-%CF%80%CE%BB%CE%B5%CF%85%CF%81%CE%B9%CE%BA%CE%AE-%CF%83%CE%BA%CE%BB%CE%AE%CF%81%CF%85%CE%BD%CF%83%CE%B7-als-amyotrophic-lateral-sc/
- http://www.disabled.gr/als-hellas-sillogos-asthenon-apo-amiotrofiki-plevriki-sklirinsi/
- www.alshellas.org
- https://www.logoskaigrafi.gr/2012/12/als.html?m=1