Article / Research Article
1Sotiria Hospital for Thoracic Diseases
Efstathios Koutsostathis,
Kerameikos Health Center,
Greece.
24 September 2023 ; 10 October 2023
Cystic fibrosis is the most common, life-threatening hereditary disease in children. It is transmitted by recessive somatic character affecting the exocrine glands. The disease causes abnormal ion transport in the epithelial tissues, causing dehydration, making mucus thick and sticky, causing mechanical obstruction in the pores and glands. The organs. The severity of the disease varies. Although an increased survival rate has been achieved in recent years, death is the final result as progressive pulmonary complications occur, which pose a serious threat to the child’s life. As the disease is chronic hospitalizations can be frequent.
Since the early 1900’s various medical articles began to be published, referring to pancreatic and respiratory dysfunctions. However, no one could speak of a specific condition because of the great diversity of the pathology of the disease. So, after many studies, it was proven that cystic fibrosis is the most widespread, fatal hereditary disease in the white race. In 1938, Dorothy Hansine Andersen described the disease for the first time and recognized its hereditary character due to mutations in the CFTR gene. In Greece, it was first described in 1949. Three years later, the discovery of electrolyte disorder in sweat by Paul Di Sant Agrese helped in the development of the sweat test as a method of diagnosis of the disease. In 1988, the first double lung transplantation was performed, and a year later the cystic fibrosis gene was isolated on a part of the long strand of chromosome 7 by the team of Lap-Chee Tsui, Dr. Francis Callins and Professor Jack Riordan. It is after 1993 that the first treatments, such as the first inhalant mucolytic, the first antibiotic, as well as the gene therapy with the CFTR regulator transfer through a mutant adenine in nasal epithelium, appear. In the 21st century, standards concerning the nutrition of cystic fibrosis patients and their care were published for the first time, and more than 2,000 gene mutations have been found.

The transmembrane conductance regulator CFTR is defective in cystic fibrosis. The protein produced is a channel that is deposited on the surface of epithelial cells and transports chloride ions and other molecules, such as bicarbonate. The gene encoding the CFTR protein is located on chromosome 7. Several mutations of this gene lead to the disease of cystic fibrosis. Since the discovery of the gene to date, over 2,500 mutations have been identified.
Cystic fibrosis is inherited in an autosomal recessive manner, with a 4% carrier rate in the general population. Because carriers are usually phenotypically healthy, this high prevalence suggests a potential survival advantage afforded by being heterozygous. Usually, a person suffering from this disease inherits one mutated gene from each parent. Thus, first degree siblings have a two thirds chance of being carriers, while second- and third-degree relatives have a 50% and 25% chance of being carriers respectively. The parents do not carry a mutated gene. Uniparental disomy (inheritance of both copies of a chromosome from one parent) is rare and should be detected diagnostically if the child has a developmental delay.
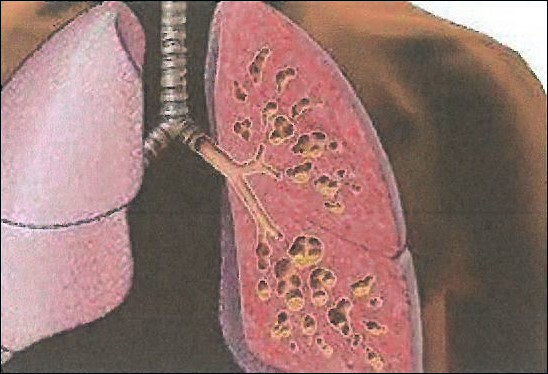
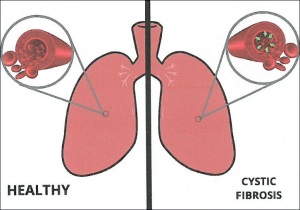
Cystic fibrosis is caused by a variety of mutations in the gene encoding a regulatory protein that controls the passage of salt (sodium chloride) across the membranes of the epithelial cells of various organs of the body such as the lungs, pancreas, sweat glands and intestine. Mutations in the gene cause reduced production or functionality of the protein and as a result in the epithelium of the affected organs a thick sticky mucus is produced, which obstructs the pores of the glands and consequently leads in the progressive destruction of the tissue of the organs (fibrosis), and their eventual failure.
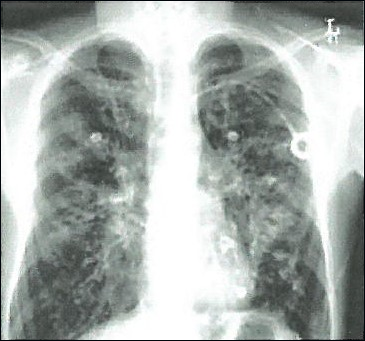
Mainly the lungs are affected, because the respiratory system of the patients is permanently colonized from an early age by the Pseudomonas and Staphylococcus microbe, resulting in respiratory failure and death. The symptoms of the respiratory system are persistent coughing, wheezing, dyspnea and repeated chest infections that cause damage to the lungs. Patients with cystic fibrosis often suffer from sinusitis, bronchitis and pneumonia and may develop nasal polyps.
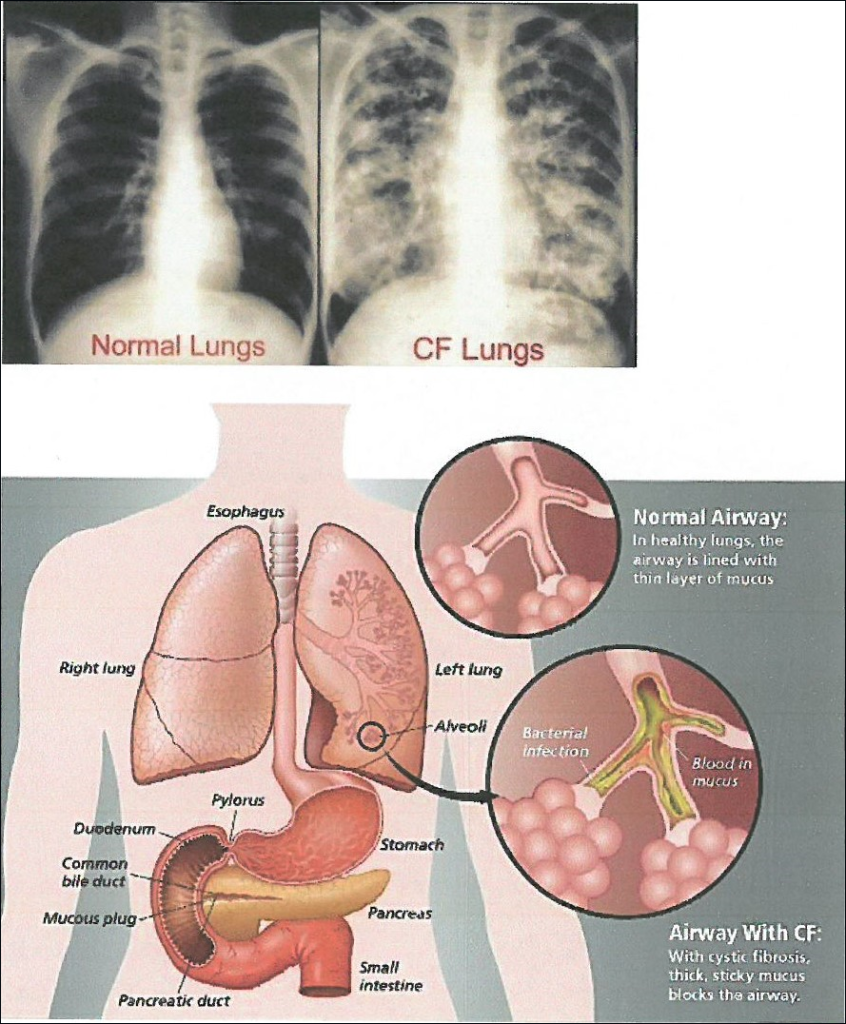
The disease affects many organs of the body such as:
- The liver by creating cirrhosis.
- The paranasal sinuses with the appearance of polyps and sinusitis from an early age.
- The bones and joints with the development of rheumatoid arthritis, osteopenia, and osteoporosis.
- The reproductive system in men, the vast majority of whom have fertility problems.
- The intestine with the development of ileus.
- The sweat glands.
The most useful laboratory test for the diagnosis of fibrocystic disease, is the measurement of chloride ion concentration by the sweat test, which is carried out in specialized laboratories.
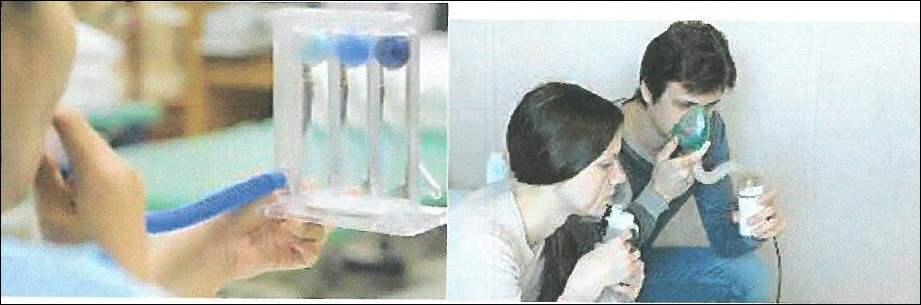
Treatment (Physiotherapy-Drugs)
The main benefits of physiotherapy to patients are improved respiratory endurance, maintenance of muscle strength, reduction of the patients’ susceptibility to various infections, treatment of these infections and maintenance of proper posture.
Physiotherapy includes various muscle strength exercises, mobility exercises of the thorax, spine, neck, and shoulders, as well as respiratory exercises.
Anti-inflammatory drugs. They can help to reduce swelling in the airways caused by recurring infections.
The doctor may prescribe mucolytics to reduce sticky mucus and loosen it. These drugs can help get the mucus out, improve lung function and prevent the worsening of lung symptoms.
Many cystic fibrosis patients take antibiotics regularly and for a long term. The antibiotics chosen, depend on the bacteria found in sputum samples. Often intravenous antibiotics are required for severe infections not controlled with antibiotic tablets.
Prenatal screening on a small blood sample can offer prospective parents the possibility for a relatively inexpensive and efficient test to identify the abnormal gene CFTR. However, the identification of cystic fibrosis mutations is more difficult in some populations of Hispanic, African origin because the nature, distribution and frequency of the mutations which cause cystic fibrosis vary considerably between populations. Disease in the fetus is tested by obtaining fetal DNA derived from chorionic villus sampling or amniocentesis biopsy.
- Book- Delmar’s Pediatric Nursing
- repository.library.teiwest.gr
- polyhoe.lib.uniwa.gr
- drtsoumpou.gr
- diaplasis.eu