









Article / Case Report
1Kerameikos Health Center, Greece
2Medical student, Athens Medical school
Efstathios Konstantinou Koutsostathis
Kerameikos Health Center
Greece
29 January 2021 ; 1 March 2021
Gaucher disease is the most common lysosomal storage disease. It is marked by deficient glucocerebrosidase enzyme activity, leading to elective accumulation of its substrate in the lysosomes of macrophages. Macrophages are most often deposited in the liver, spleen and bone marrow, creating typical symptomatology in these organs. It is a genetic disorder with autosomal recessive inheritance pattern. The extent of the damage and severity of the symptoms increase in proportion to the genetic damage in the culprit gene, GBA1. As a result, the symptoms and course of the disease may range from mild to quite acute, with potential death of the patient at a young age.
Patient, 29, was diagnosed with Gaucher disease type 1 and underwent enzyme replacement therapy, with satisfactory response. Along with the common symptoms of the disease, he had also developed the rare Erlenmeyer flask deformity.
Keywords: Gaucher Disease, Bone Involvement
Gaucher disease is a rare autosomal recessive disorder that belongs to the family of lysosomal storage diseases (LSD). These diseases make up quite a heterogeneous group of inherited diseases, which are caused by gene mutations in lysosomal enzymes or, more rarely, by mutations in lysosomal membrane protein transport. The non-functional products of the transcription of these genes prevent the normal function of the lysosome, which is a vital cellular organelle. Its main function is the digestion of non-functional, non-essential or harmful materials. To this end, its interior has an acidic pH and a large number of hydrolytic enzymes.
Specifically, Gaucher disease is characterised by mutations in the GBA1 gene, 1q21 (and rarely by deficiency in the glucocerebrosidase activator, saposin C), which produces the glucocerebrosidase enzyme (or β-cerebrosidase). This enzyme is essential for the catabolism of lipids and, more specifically, a type of fatty substances known as cerebrosides. The result of this mutation is the deficient activity of the enzyme and the consequent accumulation of cerebrosides in the macrophage cells of the reticuloendothelial system. These cells, with the so-called wrinkled tissue paper appearance, are typical of the disease and are known as “Gaucher cells”. Due to their increased presence in the spleen, bone marrow and liver, the most common symptoms of the disease are splenomegaly, bone pain and hepatomegaly, along with other accompanying symptoms (anaemia, thrombocytopenia, feeling of fatigue and haemorrhagic tendencies). In reality, the increase in the size of the infiltrated organs is due to the inflammation and fibrosis caused by the gaucher cells, and not to the volume of the cells themselves. Note that due to the diversity of genetic defects, the symptoms and severity of the disease vary, a fact that justifies the rough classification of the disease in three types: the first and most common may develop at any age and advances progressively as a chronic disease, with the symptoms mentioned above, without neurological involvement. The second type progresses rapidly from infancy, when it first develops. It is accompanied by systemic and neurological manifestations, due to the deposit of cerebrosides in the central nervous system, resulting in premature death. Lastly, the third type is clin-ically heterogeneous among the population and has the same symptoms as those of type 1. However, patients demonstrate significant neurological impairment.
The incidence of the disease is around 1/40,000 to 1/60,000 births in the general population, and 1/800 in the Ashkenazi Jewish population. The disease is diagnosed through an enzyme analysis, to determine glucocerebrosidase activity, as well as a DNA analysis. It may be managed with regular infusions of glucocerebrosidase analogues, as their administration seems to have good long-term effects (use of imiglucerase or velaglucerase). The effort to reduce the substrate of glucocerebroside (miglustat) also seems to demonstrate therapeutic benefits. Gene therapy may also be possible, as is the case in many other autosomal recessive diseases [1].
Admission Details and Diagnosis
Patient 29 presented at the Haematology Clinic of the Metaxa Cancer Hospital of Piraeus on 12 January 2015 complaining of low-grade fever in the afternoons for the last 10 days. Nothing significant arose from the family history. The clinical examination revealed lymph node blockage, hepatomegaly and splenomegaly. On the admission date, thrombocytopenia and slight anaemia were considered significant findings, following complete blood count and biochemical analysis.
Abdominal, liver and spleen ultrasound, chest and abdominal CT, and PET-CT scan were additionally requested, which were performed on the following day. These confirmed and clarified the aforementioned symptoms: lymph node blockage measuring 17x11mm in the anterior superior mediastinum and lymph node measuring 11x12mm, lymph node measuring 1×1.2cm in the right pulmonary hilum, increased liver dimensions without focal abnormality, and spleen with an anterolateral diameter of 16cm and a craniocaudal of 17cm. The antibody testing performed next was negative for toxoplasmosis and cytomegavirus, however, the mono test was positive, which possibly puz-zled the doctors momentarily. Nevertheless, it was considered negligible for someone of the patient’s age. On suspicion of a haematological disease, a myelogram was also performed, revealing marrow alterations, with the most prevalent being the presence of macrophages (histiocytes) that resembled gaucher cells. The extensive bone marrow infiltration by these unique cells was confirmed after bone marrow biopsy, which also led to testing the patient for glucocerebrosidase deficiency. A blood sample was sent to calculate the chitotriosidase biomarker (which is quite high even in presymptomatic patients and directly correlates with the disease burden). Meantime, pelvic/hip X-ray and heart ultrasound were performed, which did not reveal any significant findings, while the femoral bone/ knee MRI revealed bone restructuring abnormalities consistent with Erlenmeyer flask deformity. Given that a few days later, the chitotriosidase level was abnormally elevated (value: 4,338 nmoles/ml/hr), supplementary testing was requested for the patient and his parents. In the patient, the DNA PCR test revealed the presence of heterozygous mutations N370S and L444P. The mother’s chitotriosidase was normal, but the genetic testing detected heterozygous mutation L444P in the GBA gene, a fact that rendered the patient’s mother a carrier of Gaucher disease. As a result, the diagnosis was Gaucher disease type 1, i.e. without neurological involvement. Before the start of treatment, an associate thoracic surgeon deemed the biopsy of the pathological tissue in the mediastinum quite challenging due to its position. The patient suggested receiving treatment for Gaucher disease for the time being, with ongoing follow-up for the lymph node mass.
Course and Treatment
After diagnosis, the course of the disease was smooth.
The patient was administered treatment with imiglucerase, a medication indicated for long-term use in Gaucher disease. It acts similarly to β-cerebrosidase in the body, reducing the cerebroside burden in affected tissues, thus reducing the intensity of typical symptoms. One-and-a-half months and 4 medication doses later, bone density was normal, liver size and spleen size were normal, and complete blood count and biochemical analysis were within the normal range. Until the end of May, the patient was monitored with medical tests, without any complications. A PET-CT scan was then performed, revealing the persistent retrosternal lymph node mass, without any difference from the previous imaging scan, and, additionally, reactivation of the thymus gland. Spirometry pushed the patient to quit smoking.
The 9th dose had been infused by that time and the chitotriosidase biomarker had dropped to 2,748 nmoles/ml/hr.
Follow-up in July with a chest CT revealed residual soft tissue attached to the thymus gland, which demonstrated significant hypertrophy (possibly of a counterbalancing type). At the same time, presence of imiglucerase IgG antibodies in the patient’s plasma was negative.
At the end of September, after the 17th dose, an increase in spleen size was observed via ultrasound, which soon subsided. In mid-October, patient reported low-grade fever, which also subsided after he received the 19th dose. A PET-CT scan was performed in November to check response to treatment. Both the retrosternal mass, of soft tissue in nature, and the thymus hyperplasia remained essentially unchanged from the previous scan. However, the general consensus was Gaucher disease in remission, with normal metabolic activity of the spleen, liver and bone marrow. Around the same time, chitotriosidase testing gave a value of 1,962 nmoles/ml/hr.
The treatment continued at the same rate, with patient follow-ups for the next year.
On 22 March 2017, two years and 51 medication doses later, new spirometry was performed revealing that the 31-year-old patient had the pulmonary function of a person aged 60. On 13 October 2017, chitotriosidase was measured at 916 nmoles/ ml/hr.
This case demonstrates the coexistence of a rare disorder, Gaucher disease, with the rare Erlenmeyer flask deformity. This clinical manifestation is quite a rare and pathognomonic alteration of the disease. That is why it must be investigated in cases of unexplained splenomegaly in younger individuals.
Table of Figures
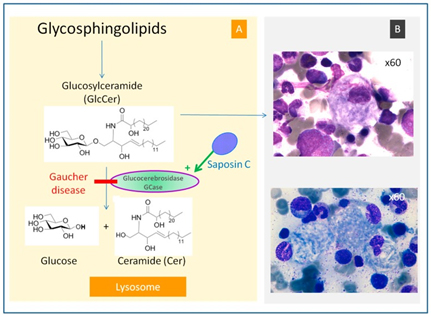
Figure 1: The typical Gaucher cell in a bone marrow biopsy[2]
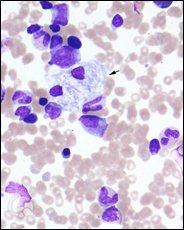 Figure 2: Hydrolysis of glucosylceramide by the glucocerebrosidase enzyme in the lysosome (A). Note that G Case is activated by saposin C. (B) Gaucher cells: they infiltrate various organs (e.g., bone marrow, spleen, and liver) and are responsible for the major signs of the disease [1].
Figure 2: Hydrolysis of glucosylceramide by the glucocerebrosidase enzyme in the lysosome (A). Note that G Case is activated by saposin C. (B) Gaucher cells: they infiltrate various organs (e.g., bone marrow, spleen, and liver) and are responsible for the major signs of the disease [1].
- Stirnemann, Jérôme et al. (2017) “A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments.” International Journal of Molecular Sciences. Vol. 18(2): 441. doi:10.3390/ijms18020441
- Lazarchick, John (2011) “Gaucher’s Disease – 1.” ImageBank, American Society of Hematology, 27 Sept. 2011, imagebank.hematology.org/image/1789/gauchers-disease–1.